

AI Is Now Treating Cancer (And This Begins An Exciting Era For Modern Medicine)
A Deep Dive Into How AI Helped Humans Discover A Novel Drug That Treats Liver Cancer

A groundbreaking research published in the Chemical Science journal has just taken humanity a step ahead in the field of modern medicine.
The team of researchers used AlphaFold2, a protein-folding AI model developed by Google DeepMind, for this discovery.
They used this model to discover a novel drug that can be used to treat Hepatocellular carcinoma (HCC), a deadly form of liver cancer that lacks effective treatments today.
This story is a deep dive into the process of this drug’s discovery from scratch.
It is divided into the following subheadings to make it easy to understand from first principles —
An Introduction To How Cancers Work
An Introduction To Developing Cancer Treatments
An Introduction To Proteins
The Challenge In Figuring Out The 3D Protein Structures
An Introduction To How The AlphaFold Models Work
A Deep Dive Into Cancer Drug Discovery Using AI
An Introduction To How Cancers Work
Cancer is a disease of abnormal cell growth in the body.
Everything starts from a single cell in the body when its genetic code is altered (Mutation).
These alterations can occur because of multiple factors —
Physical (UV rays causing skin cancer)
Chemical (Tobacco smoke causing lung cancer)
Biological (Hepatitis B virus causing liver cancer)
Inherited genetic changes (BRCA mutations in breast cancer)
These genetic alterations can occur in two ways —
Abnormal activation of growth-promoting genes (Oncogenes) in a cell
Abnormal inactivation of genes that inhibit abnormal cell growth (Tumor suppressor genes) in a cell
Either of these causes a normal cell to become cancerous.
Things happen similar to a chain reaction where abnormal cells keep surviving better than normal cells, growing unchecked, migrating to different body parts (a process called Metastasis), and taking over one’s normal bodily functions.
Think of it as if someone let one’s body cells lose, and now they are growing unchecked consuming all of the available resources and growing everywhere they find space.
When we look deeply, we notice that many of these genetic alterations/ mutations lead to abnormally functioning proteins in one’s body.
And, it is these proteins that then cause the cells to become cancerous.
Now that you have a general idea of how cancer happens, let’s move forward.
An Introduction To Developing Cancer Treatments
There are multiple ways in which cancer-treating drugs work.
One of these is by targeting the abnormal proteins present in the cancer cells.
In this process, researchers have to first figure out the 3D structures of these proteins in a lab because it is these structures that determine the biological activity of a protein.
Once determined, they then build therapeutic molecules that target these proteins.
This process is called Structure-based drug discovery (SBDD).
Unfortunately, this process of determining the 3D structures of proteins is extremely challenging.
The target proteins have to be first produced and purified and then crystallised in sufficient quantities.
These crystals are then studied using techniques such as —
X-ray crystallography, where the diffraction pattern of X-rays passed through the crystal, is studied to produce its 3D structure.

{kind=link}
Nuclear magnetic resonance (NMR) spectroscopy, where the magnetic properties of a protein’s atoms are used to determine its 3D structure
Cryo-electron microscopy, where frozen protein crystals are viewed using an Electron microscope
Once the structure is figured out, it is released in databases such as the Protein Data Bank.
This process sounds quite straightforward but in reality, protein crystallisation is a painstakingly difficult and time-consuming process.
“How difficult?”, you might ask.
It can take someone’s whole PhD to work on a single protein and there’s quite a high chance that this endeavour might fail badly.
There have even been instances where scientists have waited for decades to crystallize a protein and even then things haven’t worked at all.
(It’s tough being a biology researcher.)
Now that you know how cancer treatments are developed, it would be nice to know about proteins in a little bit more detail.
An Introduction To Proteins
Proteins are large molecules that perform extremely important biological functions in our bodies.
And, it can be confidently stated that without proteins, life as we know it would not exist.
It is the genetic material in our cells that gives rise to these bio-machines.
Chemically, proteins are composed of sub-units called amino acids.
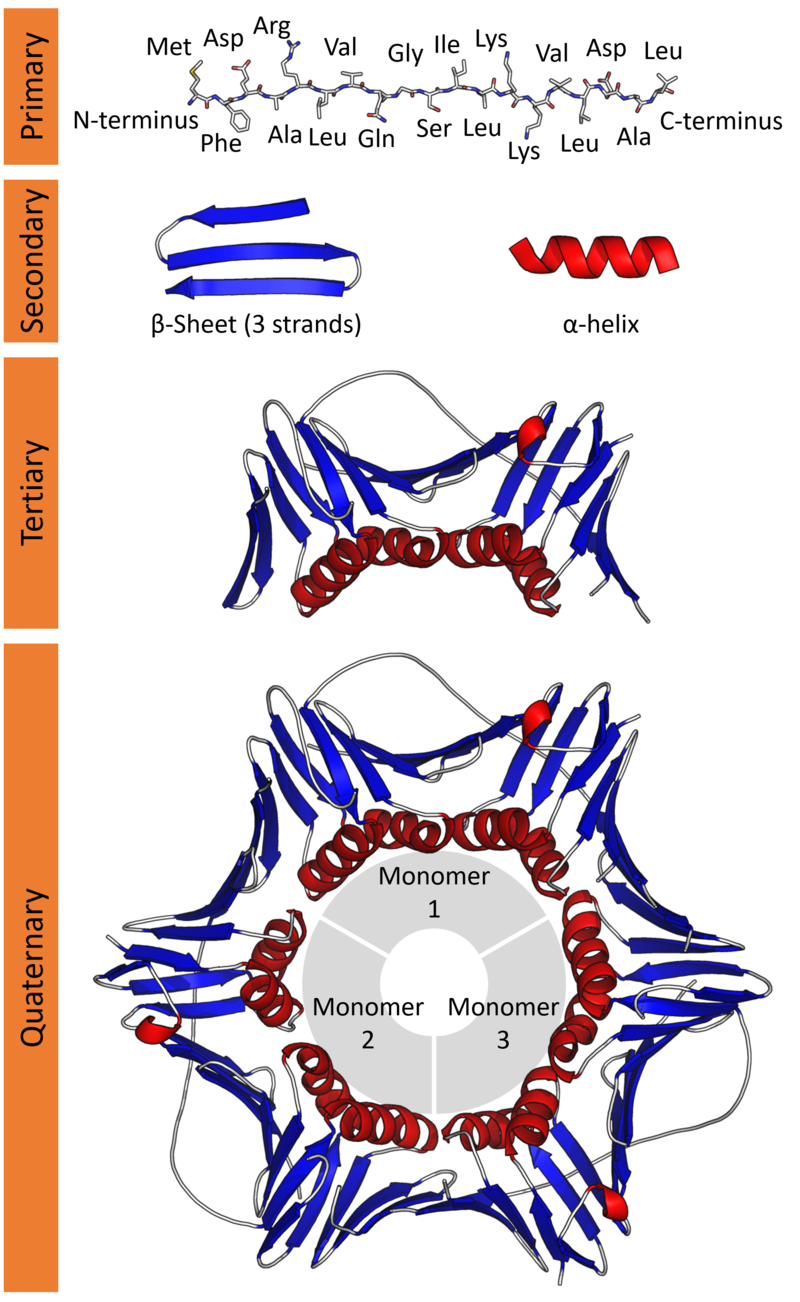
These amino acids connect together forming long chains (just like beads in a string). This is the Primary structure of a protein.
This structure folds further in helical and sheet-like appearances called α-helix and the β-sheets respectively. This is the Secondary structure of a protein.
This further folds up into a compact globular structure called the Tertiary structure of a protein.
Lastly, many Tertiary subunits can come together to form the Quarterny structure of a protein.
{kind=link}
It is this 3D Tertiary/ Quaternary structure of a protein that gives rise to its function.
Easy it might seem, but given the primary structure of a protein, it is extremely tough to predict the tertiary/ quaternary structure.
This difficulty is expressed well by Cyrus Levinthal, a molecular biologist, who published that an unfolded protein chain can have 10³⁰⁰ possible conformations.
If a protein were to reach its correctly folded structure by sequentially sampling all the possibilities, it would require a time longer than the age of the universe.
Paradoxically, most proteins fold by themselves in a time scale of microseconds to milliseconds.
The Challenge In Figuring Out The 3D Protein Structures
Biologists have long dealt with the frustrating process of figuring out the right protein structures.
To address this, the community conducts a worldwide experiment for protein structure prediction that takes place every two years.
This is called Critical Assessment of Structure Prediction (CASP).
Really smart participants are invited here to submit structures for a set of proteins for which the experimental structures are known but not yet released to the public.
Usually, the winning models of this competition have been pretty mediocre but a team of researchers at Google DeepMind with their AlphaFold AI model dramatically changed this in 2018.
They then improved their model to call it AlphaFold2 and won the competition again in 2020, with a huge margin.
In 2021, the team published their model in the Nature Journal and open-sourced its code.
By 2022, they released a public database of nearly 1 million protein structures, that is nearly all catalogued proteins known to science, all free to be accessed by anyone on the internet.
Let’s learn about these models that solved protein folding, in more detail.
An Introduction To How The AlphaFold Models Work
AlphaFold1
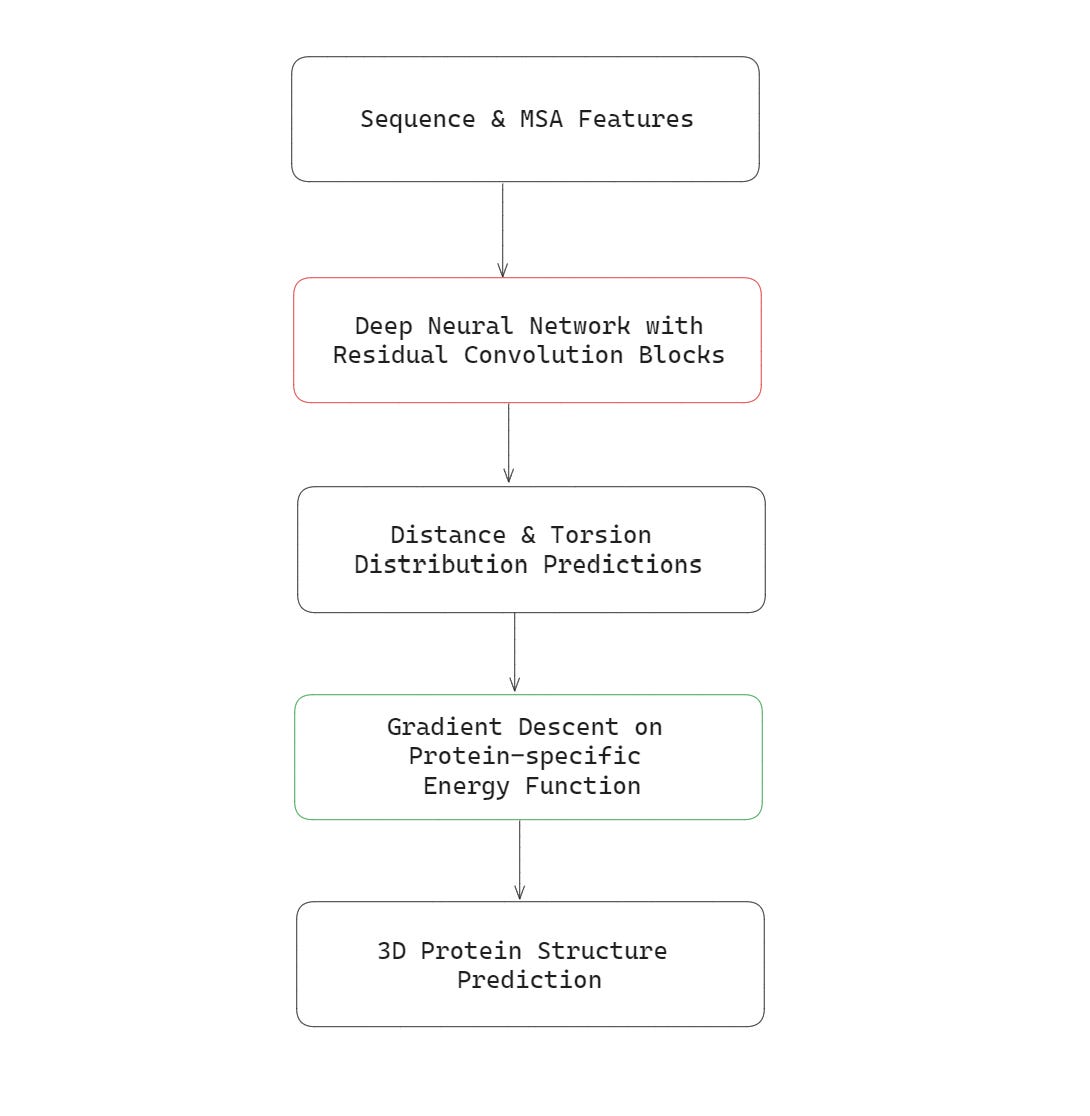
The first AlphaFold model that beat all other competitors in CASP13 in 2018, used Residual Convolutional layers in its architecture.
This model’s inputs were —
the sequence of amino acids in the primary structure of the protein to be predicted
Multiple Sequence Alignment (MSA) features
(We will soon come back to what MSA features are, in detail.)
This model’s outputs were:
the probability distribution of distances between pairs of amino acids in the protein, and
the probability distributions of torsion angles in the protein backbone
These outputs were then used to create the final 3D structure of the protein using gradient descent optimisation to minimise a protein-specific energy function.
This energy function used various physical features (such as bond lengths, bond angles etc.) to make sure that the final predicted structure had the lowest energy or the most stable conformation.
AlphaFold2
The first AlphaFold model was quite good but the DeepMind’s researchers’ team didn’t stop here.
In 2020 (at CASP 14), they presented their latest version called AlphaFold2 — which was so good at predicting protein structures that the scientific community considered the 50-year grand challenge of protein–folding problem — solved.
Let’s learn how it works.
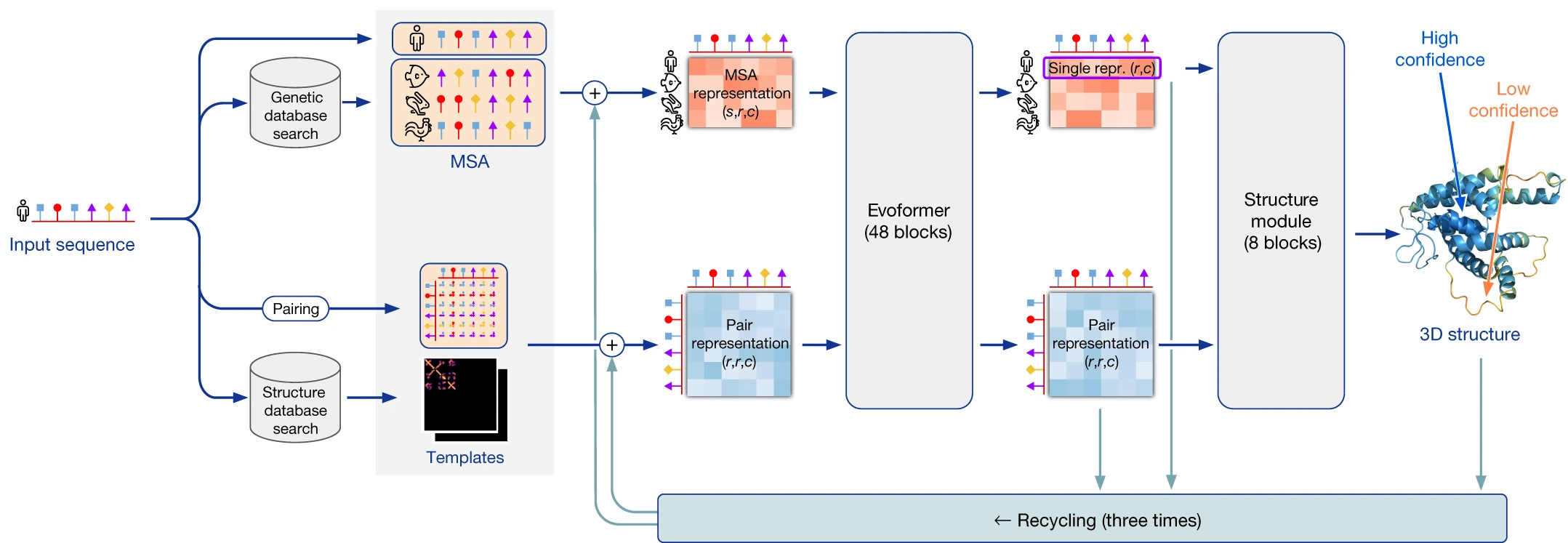
This model in comparison to the first is an end-to-end prediction model that directly outputs the 3D structure of the protein, given the inputs.
The model inputs are —
the sequence of amino acids in the primary structure of the protein to be predicted
Multiple Sequence Alignment (MSA) features
Pairwise representation between amino acids in the input sequence
Structural templates
Let’s explore these inputs.
Multiple Sequence Alignment (MSA) features
AlphaFold2 uses an important intuition from Evolution here.
This is —
Evolution preserves the function of a protein (determined by its 3D structure) while mutations occur in the primary sequence.
In other words, proteins that are evolutionary-related have very similar structures even when their primary sequences have changed over time due to mutations.
To leverage this, the input sequence is searched for in large open-source protein databases for homologous sequences.
Found sequences are then aligned together with the input sequence to create the Multiple Sequence Alignment (MSA) to be used as the input to the model.
This gives the model an idea about the evolutionary history of a protein (conserved regions, correlated mutations etc.)
Pairwise Representation
This is a matrix created by the outer product of the input sequence with itself.
This helps de-emphasise the sequential information in the input sequence and instead helps in figuring out the interactions of different amino acids with each other that might be closer to each other in the 3D structure.
Structural Templates
These are the previously known protein structures that might have a similar structure to the protein being inferred.
AlphaFold2's architecture consists of an Evoformer & a Structure module.
Evoformer
The input representations as previously described are passed to a 48-block neural network called Evoformer.
This block uses self-attention mechanisms and feed-forward networks to improve on the input representations.
An important and novel mechanism used by the Evoformer that should be highlighted here is the Triangular Attention which helps capture the geometric relationships between different amino acids.
Given 3 amino acids (A, B, C) depicted using a triangle in the protein structure, the distance AC should depend on BC and AB.
This places a constraint on the distance AC during its update in the training process.
Thus, using Triangular attention helps ensure that the model updates the predicted distance between two amino acids (A, C) by considering all the triangles that AC is a part of.
Structure Module
The outputs of the Evoformer are fed to the 8-block Structural module that helps predict the 3D coordinates of all amino acids in the protein structure.
This model uses another novel attention mechanism called Invariant Point Attention that ensures that the predicted 3D coordinates are invariant to rigid-body transformations.
This is because the absolute orientation and position of a protein in 3D space are not biologically relevant. What matters is the relative positions of the amino acids within the protein.
During the training process, the final protein structure prediction is added to the inputs of the Evoformer again 3 times.
This step is called Input Recycling and helps the model reach a better performance during the training process.
Finally, the model provides us with two prediction accuracy metrics —
p-LDDT: a per-amino acid estimate confidence score from 0–100
Predicted Aligned Error: an error in the expected position between amino acid pairs in the predicted structure
A Deep Dive Into Cancer Drug Discovery Using AI
Coming back to the cancer drug discovery, the first of its kind, here’s how this was done.
In their experiment, the researchers at Insilico Medicine targeted hepatocellular carcinoma (HCC), a type of liver cancer that is highly prevalent and lacks effective treatments.
PandaOmics
To begin with, they used PandaOmics, an AI-powered biocomputational software that uses scientific-text-derived information and OMIC data to reveal connections between genes and the disease of interest that were not apparent at first sight.
PandaOmics helped the researchers obtain a list of 20 drug targets for hepatocellular carcinoma and CDK20 (Cyclin Dependent Protein Kinase) was identified as the most promising candidate out of these.
CDK20, an enzyme protein, is found to be abundant in tumour cells of hepatocellular carcinoma and many other cancers, such as colorectal and ovarian cancers.
Thus, the researchers aimed to develop drugs that can act against this enzyme to potentially slow down the growth of this cancer.
AlphaFold2
The AlphaFold2 model, as previously described, was used to predict the 3D structure of CDK20.
Chemistry42
This predicted structure was then passed to Chemistry42, an AI-powered software that helps to predict and design novel chemical entities with desired biological properties.
Chemistry42 helped the researchers find 7 compounds of interest that could bind to CDK20 and inhibit it.
Out of these, compounds named ISM042–2–001 and ISM042–2–048 were found to be highly potent against CDK20 when further tested on hepatocellular cancer cells in the lab.
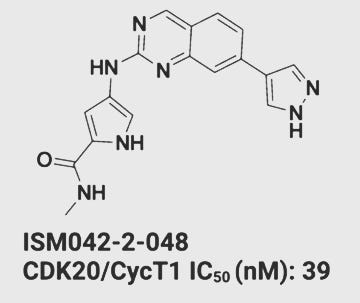
ISM042–2–048 (IC-50 is the metric used to measure drug potency) (Source: Edited image from the original research paper published in Chemical Science journal)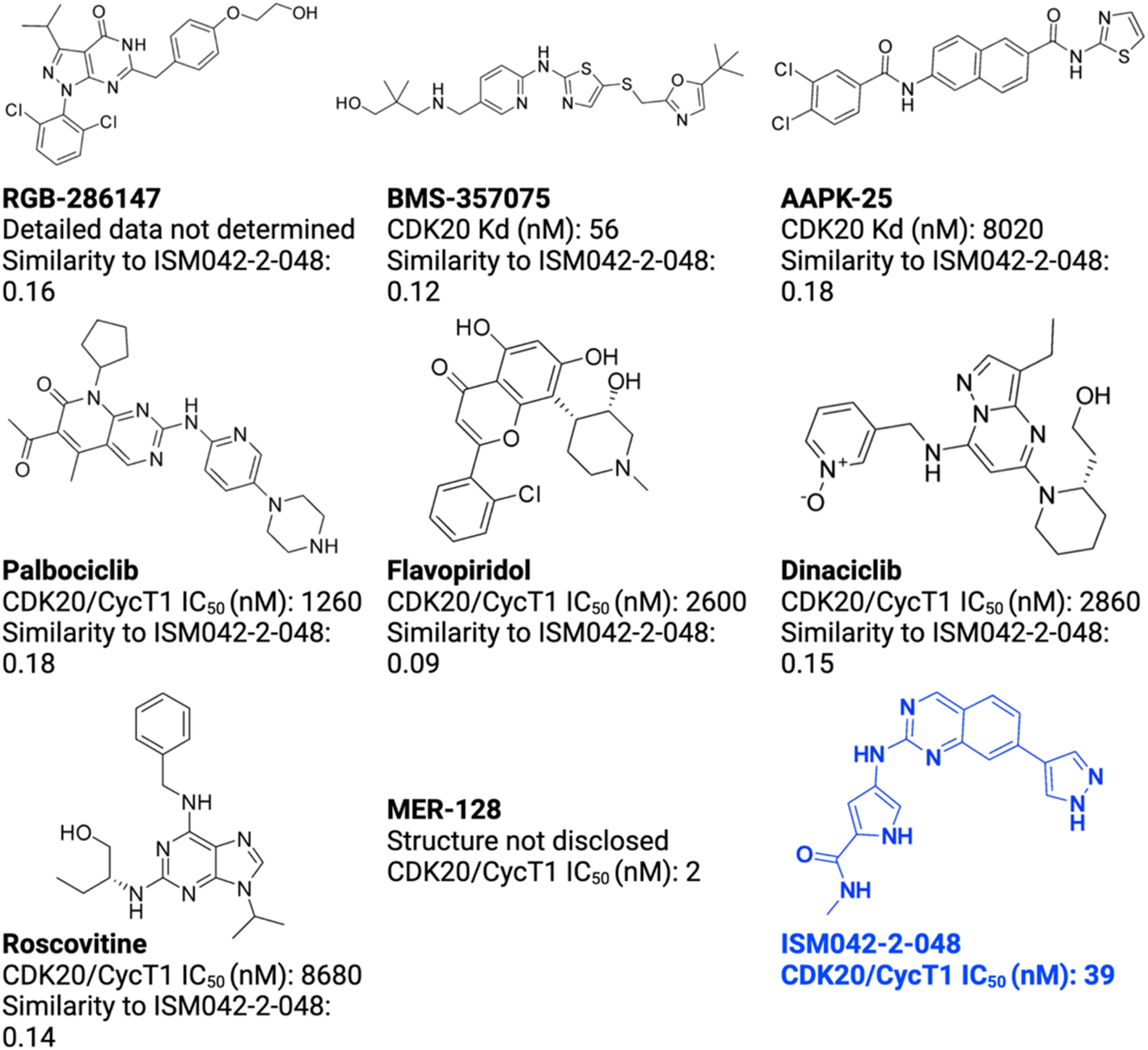{kind=link}
Surprisingly, ISM042–2–048 has a stronger effect on CDK20-overexpressing HCC cells and does not kill healthier cells indiscriminately.
This is a desired property when developing cancer therapy so that healthy cells are killed along with cancer cells.
And, this is how multiple AI models were used together to discover the world’s first cancer treatment in-silico!
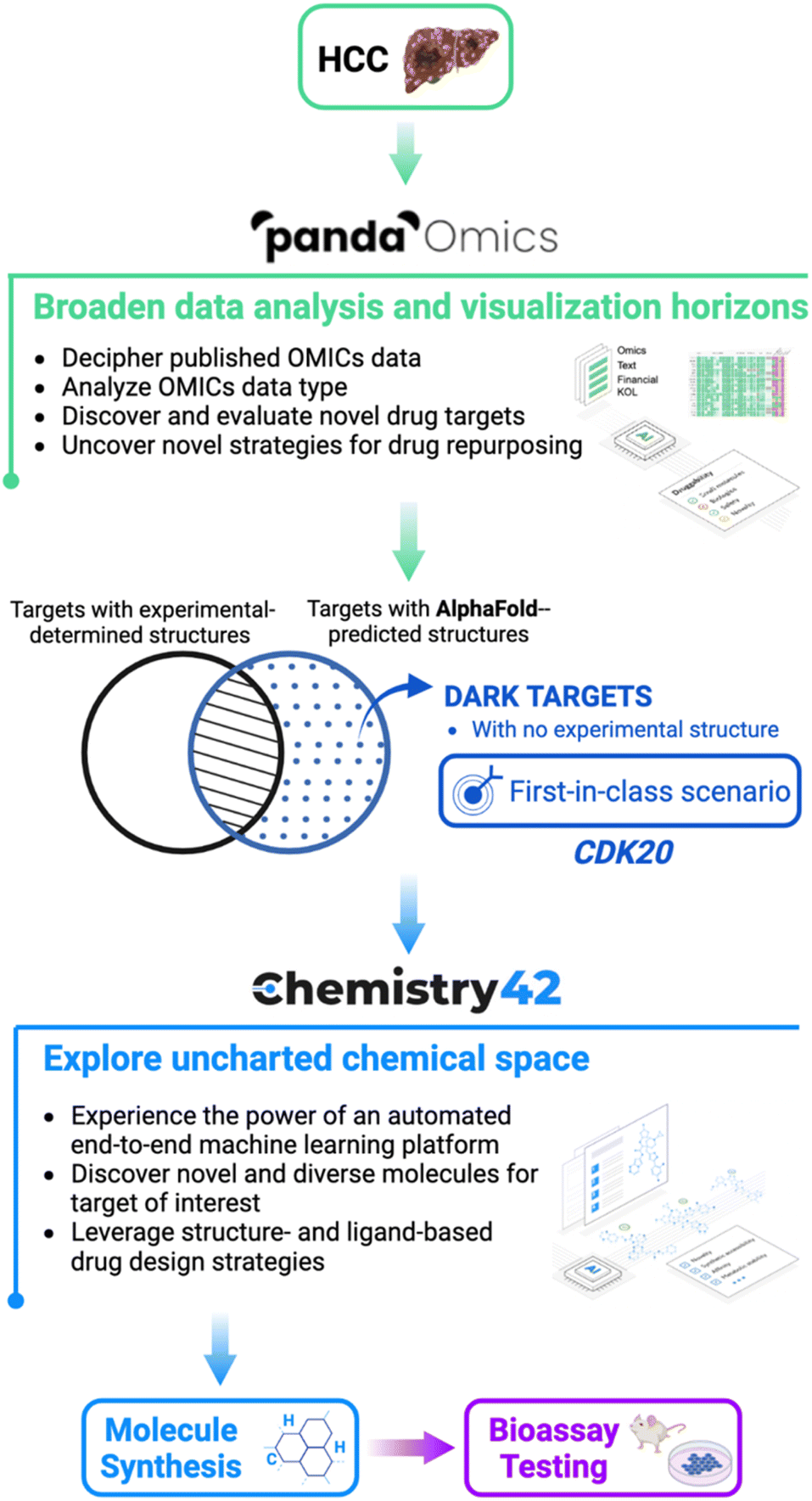
{kind=link}
Further Reading
Highly Accurate Protein Structure Prediction with AlphaFold by Simon Kohl by heidelberg.ai on Youtube
The use of AI in discovering drugs against cancer is truly remarkable!
What are some of the most impressive AI use cases that you know about?
Share them with me in the comments below!
"insilico medicine" if you can how this company work kindly share that too